(Modified from an original protocol by William C. Ratcliff, Nicholas Beerman and Tami Limberg)
Background
The evolution of multicellularity was one of the major transitions in the history of life. It allowed for an increase in complexity via the partitioning of cells for specialized functions, all working together within one entity. Multicellular organisms could move farther, grow bigger, and survive in more diverse ecosystems. Despite these advantages, we know very little about how organisms made the switch from unicellularity to multicellularity. Amazingly, we do know it happened repeatedly throughout life’s history, in at least 2 dozen independent evolutionary events. Yet how did these events occur? What were the selective benefits, if any, to the first multicellular organisms? What environmental conditions might have favored this transition? These are some of the questions that scientists are still grappling with, and that you will explore in this lab.
Fossil evidence suggests that multicellular organisms first appeared over a billion years ago. Even the most recent transition we know of happened over 200 million years ago. This makes it especially difficult to study the origin of multicellularity in a lineage, because the accumulation of so many genetic changes over this span of time makes it hard to determine which changes were important during the actual transition. While we may not be able to determine exactly how any one of these transitions occurred in the past, we can artificially recreate similar events in order to infer how these transitions might have happened.
In the paper you read for this week, Ratcliff et al. (2012) carried out a novel experiment to evolve a simple form of multicellularity in the lab, starting with single-celled microbes. The authors created an environment that favored mutant strains that formed clusters of cells (the first step in the transition to multicellularity) by subjecting Baker’s yeast (Saccharomyces cerevisiae) to daily selection for fast settling through liquid medium. Within just a few weeks, yeast that formed snowflake-shaped clusters of cells (Figure 1) evolved and displaced their single-celled ancestors. “Snowflake” yeast display several hallmarks of multicellularity, including juvenile and adult life stages, determinate growth, and a rudimentary cellular division of labor utilizing programmed cell death. Similar experiments have since been done on the single-celled alga Chlamydomonas reinhardtii, with similar results (Ratcliff et al. 2013).
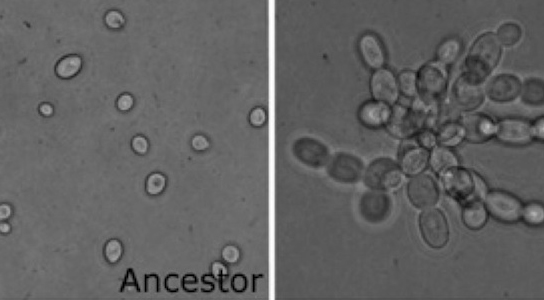
Figure 1: The Ratcliff et al. experiment started with single-celled yeast (left) and multicellular snowflake yeast (right) evolved under selection for fast settling (photo credit: Will Ratcliff)
While these experiments have demonstrated that early steps in the transition to multicellularity can be observed, selection for fast settling was probably not the primary driver of multicellularity in nature. Are there more natural environmental conditions that might have selected for multicellularity? Because the evolution of multicellularity has occurred so many times throughout evolutionary history one might hypothesize there were many different environments for which being multicellular conferred a fitness advantage. However, one could also ask why we don’t see multicellularity arise even more frequently. Why did some lineages retain their unicellularity? Again, we return to the concept of fitness tradeoffs. What are the tradeoffs of multicellularity?
Lab Overview
This lab contains two parts, both of which allow you to explore tradeoffs to being multicellular.
1. To explore the advantages of multicellularity in the face of a more “natural” selective pressure you will use unicellular and snowflake yeast to test whether predation by small-mouthed organisms (Figure 2) can select for cluster formation.
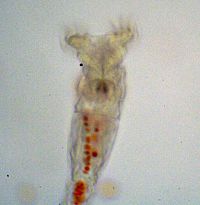
Figure 2: Rotifer after eating red-dyed unicellular yeast
2. To explore a potential disadvantage to being multicellular, you will perform a direct competition between unicellular and snowflake yeast to test whether one has a growth advantage over the other.
Lab Logistics
Day 1 (Tuesday)
Daily Overview
Initial plating of multicellular (M) and unicellular yeast (U) on YPD plates, both alone and in competition; observe rotifer predation on both unicellular and multicellular yeast
Materials
- 18mm test tube rack containing:
- 1 18mm tube of 4 ml Saccharomyces cerevisiae unicellular yeast (labeled "U")
- 1 18mm tube of 4 ml Saccharomyces cerevisiae snowflake yeast (labeled "M")
- 1 18mm sterile tube with glass beads for plating
- 3 18mm sterile tubes with 9.8 ml of YPD medium
- 1 18mm sterile tube with 3.84 ml of YPD medium (labeled "Dilute U")
- 6 Petri Dishes with YPD agar
- One 96-well microtiter plate (filled with 270µL of .86% saline in each well)
- Pipettemen and sterile tips
- 2 compound microscopes
- 2 depression slides
- 1 flat slide
- coverslips
- 1 eppendorf tube containing methylene blue dyed multicellular yeast
- 1 eppendorf tube containing Congo red stained unicellular yeast
- Beaker with 95% ethanol (for disposing the glass beads)
- 70% ethanol spray bottle & Kimwipes
- Waste container
- Burner & striker
- Sharpie markers for labeling your Petri dishes and tubes
- Gloves
Protocol
Preparing the Competition
- Put on gloves and sterilize bench with 70% ethanol. Spray ethanol on a kimwipe and use this to sterilize your pipettemen. After the alcohol evaporates, turn on gas and light burner.
- Vortex the U tube for ten seconds, then IMMEDIATELY with a sterile tip pipette 160 uL of U culture into the tube labeled "Dilute U", discard tip.
- Label an unlabeled tube with 9.8 mL YPD "Competition 1" (also write your group name and the date on this tube)
- Vortex the M tube for ten seconds, then IMMEDIATELY use a sterile tip to pipette 100 uL of the M culture into the "Competition 1" tube, discard tip.
- Vortex the "Dilute U" tube for ten seconds, then IMMEDIATELY use a new sterile tip to pipette 100 uL of the diluted U into the labeled "Competition 1" tube, discard tip.
- Label two more unlabeled tubes with 9.8 mL YPD "Competition 2" and "Competition 3" (add your group name and the date to both)
- Repeat steps 4 and 5 to initiate each of "Competition 2" and "Competition 3"
Diluting The Competition
- Label microtiter plate with “group name, date” and label columns #1, #2, and #3 with “Comp 1 t=0”, “Comp 2 t=0”, “Comp 3 t=0”, respectively.
- Vortex your first competition tube for ten seconds and proceed to next step immediately
- With a sterile tip pipette 30 uL of your competition into the first well (10^-1 dilution) of the column labeled "Comp 1 t=0" that contains 270 uL of 0.86% saline
- Using the same tip, pipette up and down 6 times in order to mix the solution, then discard tip.
- Repeat for competitions 2 and 3 into the appropriate columns.
- Perform a dilution series in each of the "Comp t=0'' columns of the microtiter plate (see Dilution Series for review) until you have reached a dilution of 10-2 (this should be the B row). Each time you pipette into a new well, pipette up and down 6 times. Change tips each time you move from one well to another.
- Put the three competition tubes into the spinner in the 30°C incubator.
Plating The Competition
- Label your 6 YPD Petri dishes as follows:
- Working near the flame, add 4-6 beads to each of your plates, making sure to flame the end of the glass bead tube before pouring.
- With a sterile pipette, add the corresponding dilution and amount (50 uL for -1 and 100 uL for -2). Note: PIPETTE UP AND DOWN 6 TIMES BEFORE PLATING.
- Shake the Petri dishes to spread out the yeast. Make sure beads are crossing across the midpoint of the dish (not just running in a circle along the edges of the dish) and make sure to rotate the dish (like a vinyl album rotates) as you are shaking. When Petri dishes appear to be mostly dry, flip them over to collect glass beads on the lid. Dump the glass beads into the beaker with ethanol and quickly recap the dish.
- Place Petri dishes agar-side up in the 30°C incubator.
Rotifer Predation There are two components to this part of the lab that can be done simultaneously by different members of your group (although each member should take a turn with both parts).
Part 1: Observing rotifer predation
- Pipette 100 uL of rotifers onto a depression slide (note: get rotifers from bottom of jar with some debris if you can).
- Invert the tube containing the blue stained multicellular yeast a few times to mix, then pipette 5 uL from this tube to the center of the depression slide (discard tip).
- Invert the tube containing the red stained Unicellular yeast a few times to mix, then pipette 5 uL (with a new tip) from this tube to the same area of the depression slide that you discarded the blue stained multicellular yeast (discard tip).
- Add coverslip and place slide on microscope stage.
- Observe rotifers feeding on the yeast, note how they are feeding and what they are eating.Where is their mouth? How are they moving? What do they do after they eat something? If you are having trouble focusing on your rotifers and yeast, raise your hand and a TA will help you.
For Bonus Points: Part 2: Quantifying rotifer predation
- Using a new depression slide, repeat steps #1-3 from Part 1 (Do NOT place a cover slip over over the depression). We recommend that you do this twice on the same slide, so that you have two areas of the slide in which to look for rotifers.
- Let the mixture sit for three minutes.
- Pipette up 20 uL of this mixture, but only expunge approximately half the liquid on to a flat slide to avoid overflow on the slide.
- Add coverslip and place slide on microscope stage.
- Observe and take photos of at least 5 individual rotifers/person (~20 per group) so that you can see the number of yeast cells in each rotifer's gut. You should be able to use the camera on your phone to do this through the microscope eyepiece. If nobody in your group has a camera phone, we will provide one for you.
- Count and record the number of blue cells and red cells in each rotifer's gut. You should count the number of dyed yeast cells in all of the photos that can be found under the Data tab if you do not have at least 16 photos to analyze.
Day 2 (Wednesday)
1-2 people/group needed, approximately 1 and ½ hours
Daily Overview
24 hour plating of multicellular (M) and unicellular yeast (U) competition on YPD plates
Materials
- 18mm test tube rack containing:
- 1 18mm sterile tube with glass beads for plating
- 6 Petri Dishes with YPD agar
- One 96-well microtiter plate (filled with 270µL of .86% saline in each well)
- Pipettemen and sterile tips
- Beaker with 95% ethanol (for disposing the glass beads)
- 70% ethanol spray bottle & Kimwipes
- Waste container
- Burner & striker
- Gloves
Protocol
Dilute t=24 Competition
- Label columns #5, #6, and #7 with “Comp 1 t=24”, “Comp 2 t=24”, “Comp 3 t=24”, respectively.
- Retrieve your three competition tubes from the spinner inside the incubator.
- Vortex your first competition tube for ten seconds and proceed to next step immediately
- With a sterile tip pipette 30 uL of your competition into the first well (10^-1 dilution) of the column labeled "Comp 1 t=24" that contains 270 uL of 0.86% saline
- Using the same tip, pipette up and down 6 times in order to mix the solution, then discard tip.
- Repeat for competitions 2 and 3 into the appropriate "t=24" columns.
- Perform a dilution series in each of the "Comp t=24'' columns of the microtiter plate (see Dilution Series for review) until you have reached a dilution of 10-4 (this should be the D row). Each time you pipette into a new well, pipette up and down 6 times. Change tips each time you move from one well to another.
Plating t=24 Competition
- Label your 6 YPD Petri dishes as follows:
- Working near the flame, add 4-6 beads to each of your 6 plates, making sure to flame the end of the glass bead tube before pouring.
- With a sterile pipette, add the corresponding dilution and amount (50 uL for -3 and 100 uL for -4). Note: PIPETTE UP AND DOWN 6 TIMES BEFORE PLATING.
- Shake the Petri dishes to spread out the yeast. Make sure beads are crossing across the midpoint of the dish (not just running in a circle along the edges of the dish) and make sure to rotate the dish (like a vinyl album rotates) as you are shaking. When Petri dishes appear to be mostly dry, flip them over to collect glass beads on the lid. Dump the glass beads into the beaker with ethanol and quickly recap the dish.
- Place Petri dishes agar-side up in the 30°C incubator.
Day 4 (Friday)
1-2 people/group needed, approximately 1/2 hour
Daily Overview
Counting of t=24 and t=0 plates''
Materials
- Sharpie markers
- Gloves
- Clicker Counter
Count t=24 and t=0 Competitions
Counting Plates
- Retrieve your six "t=0" and six t=24 competition Petri dishes from the incubator.
- Count the number of colonies on each dish (using a sharpie and clicker counter) and record the number on the dish. Blue Sharpies can be used for M colonies and red for U colonies. The colonies formed by M and U strains look different (see Figure 3).
- Enter the data into the appropriate Class Data Google Sheet, which can be found under the Data tab on the course website.
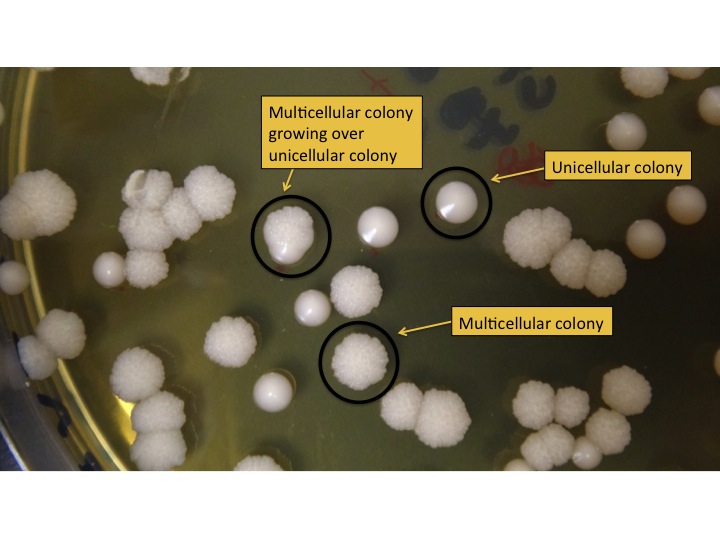
Figure 3: Unicellular and multicellular colonies on a plate. U colonies look shiny and have smooth edges. M colonies look matte and have rough edges
Putting it All together
Yeast Competition From your colony counts, you should be able to determine which type of yeast (the unicellular or multicellular) produced more colony-forming units (CFUs) when grown in competition. Calculate relative fitness, taking into account the dilutions you performed.
For Bonus Points: Rotifer Predation If you completed part 2 of the rotifer predation protocol: Calculate the proportion of multicellular yeast consumed and use a chi-square test to determine if the difference of unicellular and multicellular cells consumed by the rotifers is significant.
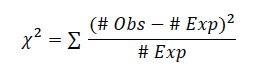
Questions
- Was one type of yeast preferentially eaten by the rotifers? Explain what might account for a feeding bias (or lack thereof).
- Report the fitnesses of your competitions, what does this pattern tell you?
- Why do we dilute the unicellular yeast down before starting our competitions?
- Do you think it is better to be a unicellular or multicellular yeast? What are the advantages and disadvantages of being either? (Your answer should include potential advantages/disadvantages that we did not directly test in this lab.) Which type do you think is more "fit" and why?
- Do you agree that the snowflake yeast is an example of a multicellular organism? If yes, what further elaborations would you expect? If no, what would need to happen to convince you that the snowflake yeast was multicellular?
Bonus Questions (.5 points each):
Part A
- Consider Ratcliff et al. (2012). If you wanted to select for yeast clusters of either 1) small, 2) large, or 3) varying size. How would you alter the experiments done in this paper? Your answer should address each of the above scenarios (1, 2, and 3).
Part B
- Consider Ratcliff et al. (2012). What additional adaptations, apart from larger cluster size, would you expect to see evidence of? Come up with at least two additional phenotypic changes and clearly articulate why you would expect these changes to occur.