Public Goods Lab
Background
What do an excreted enzyme, the scream of a ground squirrel and a contribution to public radio have in common? All of these items involve a "public good" of one kind or another. One reason that such public goods are confusing is that they seem to require that one individual provide help to others at a personal cost. A logical conclusion is that "defectors" with access to the public good (but not producing it) will have a competitive advantage over their well-intentioned neighbors. So how is it that such cooperative action could ever evolve?

Ground squirrels like this one will issue alarm calls after detecting a predator. This call informs other squirrels about nearby danger, but also might draw the attention of the predator to the caller.
To investigate this issue, we will be working with two different strains of E. coli: (1) BK27 and (2) BK26 with a plasmid (pBR) housing a gene coding for β-lactamase, an enzyme that degrades the antibiotic ampicillin. The β-lactamase enzyme is released from cells into the environment, thus it can confer a degree of resistance to other bacteria present in the growth medium even if these bacteria do not produce β-lactamase themselves. Therefore, release of β-lactamase can be viewed as a type of public good. BK27 does not produce β-lactamase but in an environment with ampicillin, it can take advantage of the β-lactamase produced by the BK26+pBR strain.
In a well-mixed environment with BK27 (e.g., a shaken tube with liquid medium), BK26+pBR, and ampicillin, the bacteria and any β-lactamase enzyme are randomly distributed throughout the environment. In such a case, BK27 experiences the protective effect of β-lactamase but pays no cost to produce it. Thus, when in competition, BK27 is expected to increase in frequency relative to BK26+pBR. When alone in an environment with ampicillin, BK27 is expected to fare poorly without its public good provider.
Can the enzyme producer ever win in competition with a defector? In a spatial soft-agar environment, bacteria grow as single-clone colonies. In this environment, the β-lactamase resource produced by BK26+pBR is more likely to benefit other BK26+pBR clones than BK27. This might give the cooperative player the edge!
In the following experiment, we will grow BK27 and BK26+pBR in pure monoculture and in competition. The competitions will take place in both a well-mixed liquid environment and a spatial agar environment. From the results of this experiment, we will determine if the production of β-lactamase is a type of public good and the conditions that could favor the evolution of microbial cooperation.
Lab Overview
In this lab, you will receive two strains of the bacterium Escherichia coli. One of these strains exports an enzyme that breaks down the antibiotic ampicillin, while the other does not. We will grow these strains alone (in pure monoculture) as well as together (in a competition) in both well-mixed and spatial environments. All of these habitats will contain ampicillin. By assessing the densities of both strains in these habitats over time, we will uncover the competitive abilities and productivies of these strains relative to one another. This information will be used to determine whether this system conforms to a standard public goods scenario.
Lab Logistics
This lab will take three days to complete. The Day 1 session (Tuesday) should take between 1 and 1.5 hours. The Day 2 session (Wednesday) will take between 1.5 and 2 hours, and it would be great to have as many group members as possible attend. The Day 3 session (Thursday) will only require one person from each group to count the Petri dishes from Day 2 (this should take approximately 1/2 hour).
Day 1 (Tuesday)
Daily Overview
Initiate competitions between BK27 and β-lactamase producing BK26+pBR in both liquid (test tube) and agar (Petri dish) habitats in the presence of ampicillin. Grow each strain alone (pure cultures) in liquid habitat with ampicillin.
Materials
- 2 TA Petri dishes (2 red stripes)
- 2 LB + Amp Petri dishes (2 black stripes)
- 18mm test tube rack containing:
- 4 18mm tubes with 5 mL LB +Amp
- 1 18mm sterile tube with glass beads for plating
- 13mm test tube rack containing:
- 1 13mm tube of 1.4 ml fully grown bacterial culture BK27
- 1 13mm tube of 1.4 ml fully grown β-lactamase producing bacterial culture BK26 +pBR
- 1 13mm tube that is empty (and sterile)
- Dry bath (at 55°C) containing:
- 2 13mm tubes with 4 mL SIB soft agar
- Eppendorf tube rack containing:
- 1 1.5 mL eppendorf tube with ampicillin stock (1 mg/mL Amp)
- One 96-well microtiter plate (filled with 270µL of .86% saline in each well)
- Pipettemen and sterile tips
- Beaker with 95% ethanol (for disposing the glass beads)
- 70% ethanol spray bottle & Kimwipes
- Waste container
- Burner & striker
- Sharpie markers for labeling your Petri dishes and tubes
- Gloves
Protocol
Prepare pure monoculture tubes:
- Put on a set of gloves.
- Label 2 of the tubes with 5 mL LB + Amp as follows:
- Sterilize bench with 70% ethanol. Spray ethanol on a kimwipe and use this to sterilize your pipettemen. After it evaporates turn on gas and light burner.
- With a sterile tip, pipette 50µL of BK27 culture from the “BK27” tube into the tube labeled “BK27 alone”. Discard tip
- Using a new pipette tip, pipette 50µL of BK26+pBR culture from the “BK26+pBR” tube into the tube labeled “BK26+pBR alone”. Discard tip.
- Vortex tubes to mix (speeds 3-5 are low enough to avoid spilling).
- Place the freshly inoculated monoculture tubes in the 37 °C shaking incubator.
Mix cultures and plate to determine densities (t=0):
- Label the 2 TA Petri dishes (dishes with 2 red stripes) as follows:
- Label microtiter plate with “group name, date” and label column #1 with “Comp t=0”.
- Label 1 empty 13mm test tube with: “Init. Mix, group name, date”
- Gently add 6-8 sterile glass beads onto both the TA Petri dishes.
- Using a sterile tip, pipette 1 mL of the BK27 culture into the tube labeled “Init. Mix”. Discard tip in waste container.
- Using a new sterile tip, pipette 1 mL of BK26+pBR culture into the same “Init. Mix” tube. Vortex to mix (make sure the vortexer is not set to too high a speed).
- Perform a dilution series in the “Comp t=0” column of the microtiter plate using the “Init. Mix” tube (Pipette 30µL from the "Init. Mix" tube into the 1st well of the microtiter plate. See Dilution Series for review) until you have reached a dilution of 10-6 (this should be in well F1).
- Using a fresh tip, pipette 100 µL of dilution 10-5 onto the center of the TA dish labeled “Comp t=0, 10-5, group name, date”.
- Repeat step 8, for the 10-6 dilution (using the dish “Comp t=0, 10-6, group name, date”)
- Shake the Petri dishes to spread out the bacteria. Make sure beads are crossing across the midpoint of the dish (not just running in a circle along the edges of the dish) and make sure to rotate the dish (like an old-fashioned vinyl album rotates) as you are shaking. When Petri dishes appear to be mostly dry, flip them over to collect glass beads on the lid. Dump the glass beads into the beaker with ethanol and quickly recap the dish.
- Place Petri dishes agar-side up in the 37 °C incubator.
Initialize liquid competitions (t=0):
- Label the 2 remaining tubes with 5 mL LB + Amp as follows:
- Add 50 µL of undiluted mixed culture from the tube labeled "Init. Mix" into the tube labeled “Liquid comp I, group name, date”
- Add 50 µL of undiluted mixed culture from the tube labeled “Init. Mix” into the tube labeled “Liquid comp II, group name, date”
- Vortex both tubes to mix (check your speed).
- Place both competition tubes in the 37 °C shaking incubator.
Initialize agar competitions (t=0):
- Label the 2 LB + Amp Petri dishes (2 black stripes) as follows:
Steps 2 through 5 must occur in less than 45 seconds, to prevent premature soft agar solidification
- Pipette 40µL from dilution 10-2 (well B1) of the “Comp t=0”column directly into the soft agar of a 13mm tube. Discard tip into waste.
- Quickly pipette 40 µL from the eppendorf tube (containing 1 mg/ml Amp) into the same 13mm tube.
- Rub the test tube between your hands to mix for a few seconds, then pour the contents of the soft agar tube onto the dish labeled “Agar comp I” and immediately (but gently) swirl the dish until the soft agar uniformly covers the surface. This should all take only seconds.
- Put your now empty 13mm tube into the waste tube rack.
- Repeat steps 2-5 for the “Agar comp II” dish.
- Let the dishes sit for 5 minutes without moving them (this will allow the soft agar to solidify).
- TURN OFF YOUR GAS
- Place your dishes in 37°C incubator, Agar-Side Down!
- Place your microtiter plate into the appropriate collection tray
- Clean up your bench and double check that your gas is turned OFF.
Day 2 (Wednesday)
2 people/group needed, approximately 1 and ½ hours
Daily Overview
Plate the results (t=24) of the pure cultures and competitions between BK27 and the β-lactamase producing BK26+pBR. Count colonies to determine the initial densities (t=0) of the strains in competition.
Materials
- 12 TA Petri dishes (2 red stripes)
- 6 MA Petri dishes (1 red stripe)
- 18mm tube rack containing:
- Sterile 18 mm tube with glass beads
- 2 15mL centrifuge tubes with 8mL saline
- 1 Spatula
- Beaker with ethanol for sterilizing spatula
- Beaker with 95% ethanol (for disposing the glass beads)
- 70% ethanol spray bottle & Kimwipes
- Pipettemen and sterile tips
- Waste
- Burner & striker
- Sharpie markers for labeling
- Gloves
- Clicker counters
Protocol
- Bring the following to your bench with your group’s name:
- Label columns 7-12 of your microtiter plate as follows:
Determine final densities in agar competitions (Part I):
- Label 2 centrifuge tubes with 8mL saline
- Label 4 TA dishes (2 red stripes):
- Label on 6 MA dishes (1 red stripe):
- Sterilize bench with 70% ethanol. Spray ethanol on a kimwipe and use this to sterilize your pipettemen. After it evaporates turn on gas and light burner.
- Add 6-8 sterile beads to each Petri dish.
- Place the spatula (the entire tip) in the beaker with ethanol so the metal part is coated (but not dripping) in ethanol. With the metal part pointing down so that the ethanol does not run back towards you, flame the metal part and let cool. Make sure the flame is completely out, then repeat.
- Loosen the cap on the centrifuge tube labeled "Agar comp I". Scrape the soft agar top layer from your "Agar comp I" dish and swirl it into the saline within the tube. You can lay the dish lid on the table. Gently hold your spatula at an angle, not perpendicular to the plate to avoid cutting into the hard agar base. You do not need to apply any real pressure, just let it limply scrape across the top of the plate. (When placing the soft agar into the centrifuge tube, hold the tube at an angle over the Petri dish just in case anything falls off the spatula).
- Continue to scrape the dish until the soft agar layer has been removed (this does not have to be perfect).
- Wipe the spatula clean with a kimwipe and sterilize the spatula (see step 6).
- Repeat the steps for your dish “Agar comp II” into the centrifuge tube labeled “Agar comp II”.
- Make sure the centrifuge caps are tightly screwed on and then vortex both centrifuge tubes at full speed for 1 minute to separate the bacteria from the agar. The agar will need 20-30 minutes to settle.
- While agar is settling move on to plate your monocultures and liquid competitions.
Plate to determine final densities in monocultures:
- Label 4 TA Petri dishes (2 red stripes):
- Add 6-8 sterile glass beads to dishes.
- Perform a dilution series in the “BK27 alone (t=24)” column of the microtiter plate using the “BK27 alone” tube (see Dilution Series for review) until you have reached a dilution of 10-2.
- Perform a dilution series in the “BK26+pBR alone (t=24)” column of the microtiter plate using the “BK26+pBR alone” tube until you have reached a dilution of 10-7.
- Using a sterile tip, carefully pipette 100µL from the 10-1 dilution of the “BK27 alone (t=24)” column onto the center of the “BK27 alone (t=24), 10-1" dish.
- Using a sterile tip, carefully pipette 100µL from the 10-2 dilution of the “BK27 alone (t=24)” column onto the center of the “BK27 alone (t=24), 10-2” dish.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- Using a sterile tip, carefully pipette 100µL from the 10-6 dilution of the “BK26+pBR alone (t=24)” column onto the center of the “BK26+pBR alone (t=24), 10-6” dish.
- Using a sterile tip, carefully pipette 100µL from the 10-7 dilution of the “BK26+pBR alone (t=24)” column onto the center of the “BK26+pBR alone (t=24), 10-7” dish.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- Place Petri dishes agar-side up in the 37°C incubator.
Plate to determine final densities in liquid competitions:
- Label 4 TA Petri dishes (2 red stripes):
- Place 6-8 sterile glass beads to dishes.
- Perform a dilution series in the “Liquid comp I (t=24)” column of the microtiter plate using the “Liquid comp I” tube until you have reached a dilution of 10-7.
- Perform a dilution series in the “Liquid comp II (t=24)” column of the microtiter plate using the “Liquid comp II” tube until you have reached a dilution of 10-7.
- From the “Liquid comp I (t=24)” column, pipette 100µL from the 10-6 dilution onto the dish labeled “Liquid comp I (t=24), 10-6 ”.
- From the “Liquid comp I (t=24)” column, pipette 100µL from the 10-7 dilution onto the dish labeled “Liquid comp I (t=24), 10-7”.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- From the “Liquid comp II (t=24)” column, pipette 100µL from the 10-6 dilution onto the dish labeled “Liquid comp II (t=24),10-6”.
- From the “Liquid comp II (t=24)” column, pipette 100µL from the 10-7 dilution onto the dish labeled “Liquid comp II (t=24), 10-7”.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- Place Petri dishes agar-side up in the 37 °C incubator.
Plate to determine final densities in agar competitions (Part II):
Now that the centrifuge tubes have settled you should see a level of foam on the top and directly below that should be a slightly clear layer and lastly a cloudy layer. You will want to pipette from the clear layer beneath the foam.
- Perform a dilution series in the “Agar comp I (t=24)” column of the microtiter plate using the “Agar comp I” centrifuge tube until you have reached a dilution of 10-7. (Note: The first dilution step can be tricky, because the agar in the centrifuge tube can clog the tip if you are pulling from a layer too deep in the centrifuge tube. If this happens, discard tip, ethanol the pipetteman, and try again.)
- Perform a dilution series in the “Agar comp II (t=24)” column of the microtiter plate using the “Agar comp II” centrifuge tube until you have reached a dilution of 10-7.
- From the “Agar comp I (t=24)” column, pipette 100µL from the 10-6dilution onto the dish labeled “Agar comp I (t=24), 10-6 ”.
- From the “Agar comp I (t=24)” column, pipette 100µL from the 10-7 dilution onto the dish labeled “Agar comp I (t=24), 10-7 ”.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- From the “Agar comp II (t=24)” column, pipette 100µL from the 10-6 dilution onto the dish labeled “Agar comp II (t=24), 10-6 ”.
- From the “Agar comp II (t=24)” column, pipette 100µL from the 10-7 dilution onto the dish labeled “Agar comp II (t=24), 10-7 ”.
- Shake dishes to spread the aliquots and dispose of the glass beads in the beaker with ethanol.
- Now repeat the above plating process with the 6 MA dishes (which will give counts for only BK27). Plate 3 dilutions (10-3, 10-4, 10-5) using columns “Agar comp I (t=24)” and “Agar comp II (t=24)” of the microtiter plate that you just used.
- Place all Petri dishes agar-side up in the 37 °C incubator.
Count t=0 initial plates:
- Count the number of colonies on each “Comp t=0” dish (using a sharpie and clicker counter) and record the number on the dish. BK27 can use arabinose, thus, the BK27 colonies will appear large and whitish-pink. The BK26+pBR strain cannot use arabinose, thus, BK26+pBR colonies are small and red. (Thus, on each dish you should have two numbers for the BK27 and BK26+pBR colonies).
- Enter the data into the appropriate Class Data Google Sheet, which can be found under the Data tab on the course website. First, you will need to find your group's name in the appropriate lab section area (please do this in the "Liquid Competitions" sheet, the "Agar Competitions" sheet, and the "Monocultures" sheet-- tabs for each sheet are located at the bottom-left of the webpage). Enter your t=0 counts in both the "Liquid Competitions" sheet and the "Agar Competitions" sheet, which means you will enter the same counts for BK26+pBR (red) colonies and BK27 (pink) colonies twice (once on each of these two sheets).
Day 3 (Thursday)
1 person/group needed, approximately ½ hour
Daily Overview
Count colonies to determine the final densities (t=24) of the strains in competition.
Materials
- Clicker counters
- Gloves
- Sharpie marker
Protocol
- Bring the following to your bench with your group’s name from stationary incubator:
- Count the number of colonies on each dish (using a sharpie and clicker counter) and record the number on the dish. The TA plates will distinguish BK27 from BK26+pBR. BK27 can use arabinose, thus, the BK27 colonies will appear large and whitish-pink. The BK26+pBR strain cannot use arabinose, thus, BK26+pBR colonies are small and red. Since “MA” stands for “minimal arabinose” (arabinose is the only carbon source), only BK27 will form colonies on these dishes.
- Enter the data into the appropriate Class Data Google Sheet, which can be found under the Data tab on the course website. Note, your lab mates should have already entered your lab group's name and inputed some plate counts from yesterday (t=0). Please note that there are three sheets, one for "Liquid Competitions," one for "Agar Competitions," and one for "Monocultures" (tabs for each sheet are located at the bottom-left of the webpage). Enter your t=24 counts in the correct sheet.
Putting it All together
From the colony counts that you have gathered, you should be able to determine whether this system conforms to a standard public goods scenario. The BK26+pBR strain is the public good producer, while the BK27 strain is the "defector." When grown alone with ampicillin, the producer is predicted to fare better than the defector. However, when grown together in a well-mixed environment, the producer is expected to suffer in competition against the defector.
Taking account of the dilutions you performed, you should be able to figure out how many cells of each strain were in the tubes when grown alone. Further, you can determine the number of cells of each strain in the competitions (tubes or dishes) at the beginning and end of the assay. Focusing on the competitions, let the number of producer and defector cells at the beginning of the assay be given by Pb and Db, respectively, and the number of producer and defector cells at the end of the experiment be given by Pe and De, respectively. The fitness of producer strains relative to the defector is given by:
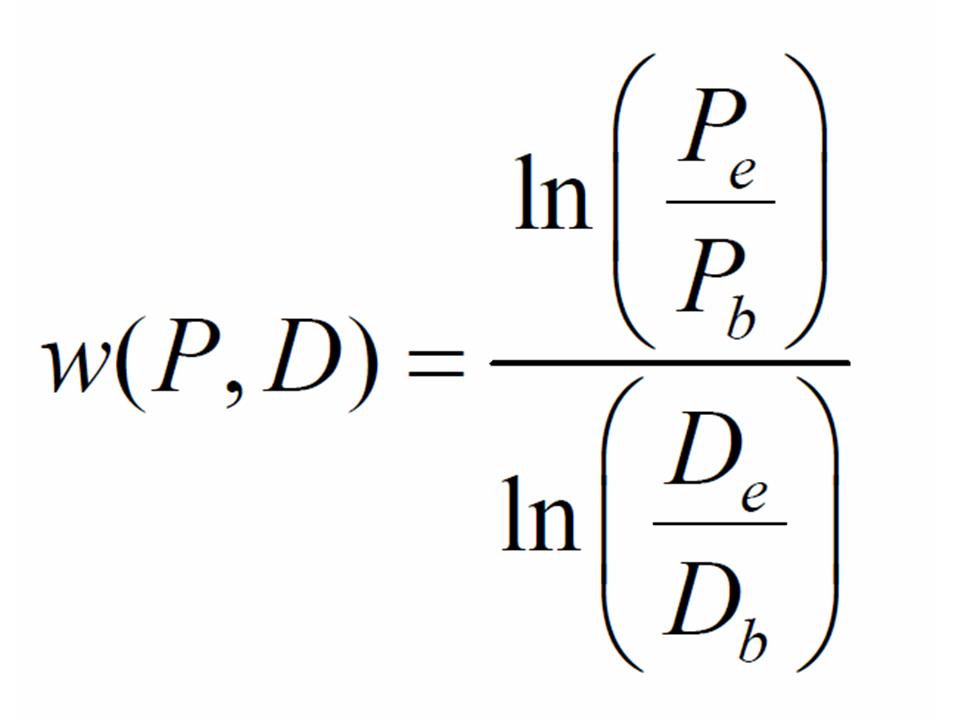
You can now compute the fitnesses of the producer relative to the defector in both liquid and agar habitats.
Questions
- Report all fitnesses for your competitions. What does this pattern suggest?
- What results might you expect in both the liquid competition and agar competitions if you added a higher concentration of ampicillin? What do you predict would happen, and why, if you decreased the ampicillin concentration?
- Imagine you changed the density of cells to have more or less of the BK26 +pBR. How might this affect growth and competition between the strains? How might fluctuating population size affect the evolution of cooperation?
- Costly production of goods that are publicly available puts the producer at a potential disadvantage. What if the system is privatized? The β-lactamase enzyme is released into the environment, but what would be the result of this lab if the enzyme was completely maintained within the periplasm space (space between the inner cytoplasmic membrane and the external outer membrane of gram-negative bacteria such as E.coli)?
- Incidentally, it seems that the actual situation for E. coli is somewhere in between "completely public" and "completely private" goods. Specifically, much of the activity of the β-lactamase occurs in the periplasmic space, but some of this enzyme "leaks out", thus, providing a public service.
- Take this experiment out of the lab and imagine its consequences within the human gut. If E.coli are releasing β-lactamase that can potentially aid the survival of different types of bacteria that would otherwise be ampicillin-sensitive, how could this affect human health and the treatment of disease?
Bonus: Studies of cooperation have traditionally focused on games such as the “prisoner’s dilemma”, where players choose between two strategies: cooperation and defection. How can you explain the long-term coexistence of two different strategies (cooperators and defectors)? You might think about the following things as you answer:
- How does the definition of cooperation (think about the payoff matrix) affect coexistence?
- How does spatial structure (think about your agar dishes) affect coexistence?