Trade-off Lab
Background
This lab is intended to illustrate a tradeoff that the bacterium Escherichia coli faces. Normally, this microbe has flagella that it uses to move in its natural environment (which includes, among other places, the mammalian gut). This flagella phenotype is costly, in terms of the protein resources to build the flagella and the energy expenditure (e.g., ATP) to turn the flagellum motor. In environments where movement is not favored (e.g., well-mixed laboratory environments, such as a shaken flask with liquid broth) E. coli strains frequently delete a portion of the collection of genes coding for the flagellum apparatus (e.g., see Zhong et al. 2003), thus eschewing the costs of flagella production and operation. In this lab, we attempt to demonstrate that a flagellated bacterium has an advantage in a spatially structured environment, while its unflagellated equivalent has the upper hand in a well-mixed environment. In a very real sense there is a tradeoff between competition for resources at a given spot (where the unflagellated strain shines) and movement to spots where there are unexploited resources (where the flagellated strain cashes in). This has been termed a competition-colonization tradeoff within ecology.
Lab Overview
In this lab, you will place the flagellated and non-flagellated strains in direct competition for resources in two different arenas: a Petri dish filled with soft agar and a shaken test tube filled with liquid broth. By estimating the initial densities of each strain as they start the competition and estimating the final densities of each strain after the end of the competition, you will be able to compute the fitness of one strain relative to the other. The hypothesis being tested is that the flagellated strain has a higher relative fitness in the spatially-structured environment (the agar-filled Petri dish), while the non-flagellated strain has an advantage in the well-mixed environment (the shaken tube).
Lab Logistics
This lab will take three days to complete: Tuesday, Wednesday, and Thursday. The Tuesday session should take between 1 and 1.5 hours. The Wednesday session only requires two members of the group to attend and should take between 1 and 1.5 hours. The Thursday session only requires one member of the group to attend and should take between 15 and 30 minutes.
Day 1 (Tuesday)
Daily Overview
To mix together the flagellated (F) and non-flagellated (N) bacterial strains and place them into a spatially structured competition arena (an agar-filled Dish) and a well-mixed competition arena (a liquid-filled Tube). Three experimental replicates (Reps) are executed.
Materials
- One culture tube marked F with 5mL of the flagellated E. coli strain
- One culture tube marked N with 5mL of the non-flagellated E. coli strain
- A tube rack containing three empty tubes and three media-filled tubes (5mL Tryptone)
- 6 test tubes filled with super soft agar (in the oven)
- A vortexer (for quickly mixing cultures)
- A 96-well microtiter plate filled with 270µL/well of saline (for diluting)
- Six empty Petri dishes
- Pipettemen and tips
- Trash bin for used tips
- A burner and a striker
Protocol
- Your TAs will start lab with a review of pipetting, pouring soft agar, and basic sterile technique.
- Label your three empty tubes Rep I, Rep II, and Rep III. Label your three media-filled tubes Tube I, Tube II, and Tube III. Label your six Petri dishes Dish I, Dish II, Dish III, Extra I, Extra II, and Extra III (place each label on the bottom of each Petri dish). Write your groups’ initials and date on the media-filled tubes and all the Petri dishes. Label your microtiter plate with your group’s initials, the date, and what will be in each column (see below).
- Turn on your gas and light your burner.
- Using a sterile tip, pipette 1000µL of the F culture into the empty mixing tube labeled Rep I and dispose of tip. Using a new sterile tip, pipette 1000 µL of the N culture into the tube labeled Rep I and dispose of tip. Vortex the mixture for 10 seconds.
- Using a new sterile tip for each dilution step, perform a dilution series of the mixture in the Rep I tube in column #1 of the microtiter plate to 10-7. Dispose of each tip after each step.
- Repeat steps 4 and 5 for the Rep II tube (diluting into column #2 of the microtiter plate) and Rep III (diluting into column #3 of the microtiter plate).
- Write your group’s initials on the Petri dish labeled Dish I. Using a sterile tip, pipette 100µL from the well with a 10-6 dilution of the Rep I mixture into a tube with 12.5mL of supersoft agar (this tube will be in the oven). Mix the agar by rolling the tube gently between your hands for a few seconds. Pour the agar into the Petri dish labeled Dish I. Immediately after pouring, gently swirl the Petri dish so that the supersoft agar is uniformly distributed across the bottom. Let this dish sit untouched for 30 minutes (this will allow the agar to solidify).
- Repeat step 7 for the 10-6 dilution of Rep II (pouring into the Dish II Petri dish) and the 10-6 dilution of Rep III (pouring into the Dish III Petri dish).
- Write your group’s initials on the Petri dish labeled Extra I. Using a sterile tip, pipette 100µL from the well with a 10-7 dilution of the Rep I mixture into a tube with 12.5 mL of supersoft agar. Mix the agar by rolling the tube gently between your hands for a few seconds. Pour into the Petri dish labeled Extra I. Immediately after pouring, gently swirl the Petri dish so that the supersoft agar is uniformly distributed across the bottom and let this dish sit untouched for 30 minutes.
- Repeat step 9 for the 10-7 dilution of Rep II (pouring into the Extra II Petri dish) and the 10-7 dilution of Rep III (pouring into the Extra III Petri dish).
- Write your group’s initials on the fresh media tube labeled Tube I. Using a sterile tip, pipette 50µL from the well with a 10-6 dilution of the Rep I mixture into the media-filled tube labeled Tube I.
- Repeat step 11 for the 10-6 dilution of Rep II (pipetting into Tube II) and the 10-6 dilution of Rep III (pipetting into Tube III).
- TURN OFF GAS. Clean up lab bench. The TAs will take your plates and tubes to the incubators (Note: these plates should not be inverted due to the supersoft agar).
Day 2 (Wednesday)
Daily Overview
To count colonies giving the initial densities of flagellated and non-flagellated bacterial strains. To plate the final outcomes of the Dish and Tube competitions.
Materials
- Six Petri dishes from yesterday, each labeled “Dish” or “Extra” (with bacteria in soft agar)
- Three culture tubes from yesterday, each labeled “Tube” (with bacteria in liquid media)
- 12 test tubes filled with super soft agar (in the oven)
- The 96-well microtiter plate from yesterday (filled with saline for diluting)
- A tube rack with three conical tubes filled with 12.5mL saline
- Twelve empty Petri dishes
- A spatula
- A foil-covered beaker of ethanol (NOTE: ETHANOL IS FLAMMABLE)
- Pipettemen and tips
- Trash bin for used tips
- Vortexer (for quickly shaking conical tubes)
- A box of Kim-wipe tissues
- A burner and a striker
Protocol
Protocol:
- With help from a TA, count the number of expanded (e.g., originating from a flagellated cell) and contracted (e.g., originating from a non-flagellated cell) colonies on the Petri dishes labeled Dish I, Dish II, Dish III, Extra I, Extra II, and Extra III. Input your data into the class computer.
- Turn on your gas and light your burner.
- At a good distance from the flame, dip the spoon end of your spatula in the beaker with ethanol and then flame sterilize this end of the spatula.
- After a few seconds of cooling, use the spatula to scrape the contents of the Petri dish labeled Dish I into the conical tube labeled Dil I. Wipe the spatula clean with a Kim-wipe tissue.
- Tighten the cap on the conical tube and vortex for 30 seconds at the highest speed setting.
- Repeat steps 3-5, scraping Dish II into conical tube Dil II, and Dish III into tube Dil III. Turn the vortex back to a speed of “6” after you finish the vortexing steps.
- Using a new sterile tip for each step, perform a dilution series of the mixture in the Tube I tube in column #7 of the microtiter plate to 10-7. Dispose of each tip after each step.
- Repeat step 7 for the
- the Tube II tube (diluting into column #8 of the microtiter plate)
- the Tube III tube (diluting into column #9 of the microtiter plate)
- the Dil I tube (diluting into column #10 of the microtiter plate)
- the Dil II tube (diluting into column #11 of the microtiter plate)
- the Dil III tube (diluting into column #12 of the microtiter plate)
- Write your group’s initials on the Petri dish labeled Final Tube I (10-6). Using a sterile tip, pipette 100µL from the well with a 10-6 dilution of the Tube I culture into a tube with soft agar (this tube will be in the oven). Mix the agar by rolling the tube gently between your hands for a few seconds. Pour the agar into the Petri dish labeled Final Tube I (10-6). Immediately after pouring, gently shake the Petri dish so that the soft agar is uniformly distributed across the bottom. Let this dish sit untouched for 30 minutes (this will allow the agar to solidify).
- Repeat step 9 for
- the 10-7 dilution of the Tube I culture (pouring into the Final Tube I (10-7) Petri dish)
- the 10-6 dilution of the Tube II culture (pouring into the Final Tube II (10-6) Petri dish)
- the 10-7 dilution of the Tube II culture (pouring into the Final Tube II (10-7) Petri dish)
- the 10-6 dilution of the Tube III culture (pouring into the Final Tube III (10-6) Petri dish)
- the 10-7 dilution of the Tube III culture (pouring into the Final Tube III (10-7) Petri dish)
- the 10-6 dilution of the Dil I culture (pouring into the Final Dish I (10-6) Petri dish)
- the 10-7 dilution of the Dil I culture (pouring into the Final Dish I (10-7) Petri dish)
- the 10-6 dilution of the Dil II culture (pouring into the Final Dish II (10-6) Petri dish)
- the 10-7 dilution of the Dil II culture (pouring into the Final Dish II (10-7) Petri dish)
- the 10-6 dilution of the Dil III culture (pouring into the Final Dish III (10-6) Petri dish)
- the 10-7 dilution of the Dil III culture (pouring into the Final Dish III (10-7) Petri dish)
- TURN OFF GAS. Clean up lab bench. The TAs will take your plates to the incubators.
Day 3 (Thursday)
Daily Overview
To count colonies giving the final densities of flagellated and non-flagellated bacterial strains from the different competitions.
Materials
- Twelve Petri dishes from yesterday, each labeled “Final Dish” or “Final Tube” (with bacteria in soft agar)
Protocol
- With help from a TA, count the number of expanded (e.g., originating from a flagellated cell) and contracted (e.g., originating from a non-flagellated cell) colonies in each Final Tube and Final Dish Petri dish. Input your data into the class computer.
Putting it all together
At this point you have a bunch of colony counts from various Petri dishes. Remember that each colony originates from a single cell. Taking account of the dilutions you performed, you should be able to figure out how many cells of each strain were in the competition arena (whether this is a tube or a Petri dish) at the beginning of the experiment and at the end of the experiment. (Don’t forget the half dilution from the saline and agar in the conical vials). Let the number of flagellated and non-flagellated cells at the beginning of the experiment be given by Fb and Nb, respectively, and the number of flagellated and non-flagellated cells at the end of the experiment be given by Fe and Ne, respectively. Note that, because of the dilutions you performed, these numbers are not always equivalent to the numbers of colonies you counted! The fitness of flagellated cells relative to non-flagellated cells is given by:
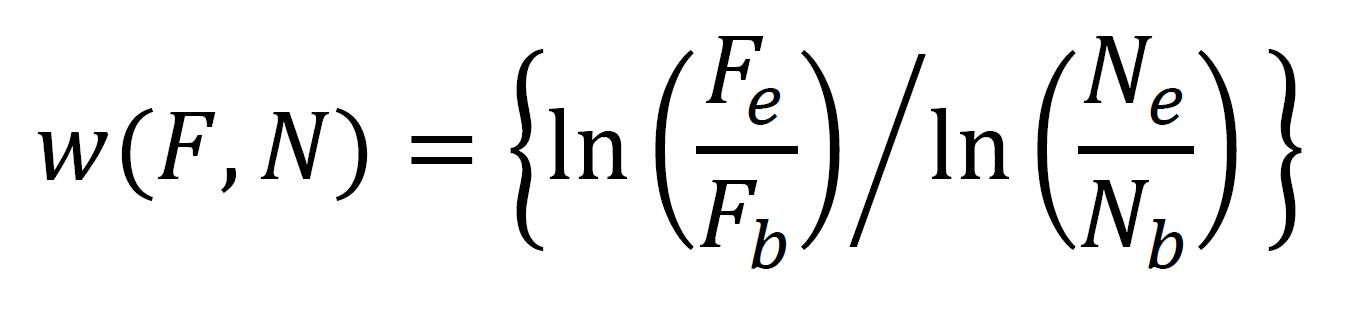
You can now compute the relative fitnesses of the strains in each of the two environments.
Questions
- What does it mean to have a relative fitness w(F,N) larger than one? Less than one? Equal to one?
- What was the fitness you computed in each environment (the tube and the Petri dish)? What explains the difference? Is there enough data to say something about the significance of these differences? Would there be enough data if all the class pooled its results?
- Is it possible to compute a negative value for fitness? If so, what would that mean?
- On the assay Petri dishes, why is there cell-free zones around the contracted colonies originating from the non-flagellated cells (i.e., where the flagellated colony seems almost to avoid the unflagellated colony)?
- Why would natural isolates of E. coli (and other bacteria) often be flagellated?
Bonus Question: Disturbance is a ubiquitous occurrence in many ecological systems. Most generally, we can think of disturbance as the removal of individuals (usually “cleared” from a given area; for example, think of forest fires or hurricanes). There is a famous hypothesis that suggests that diversity of species is highest in regions with intermediate levels of disturbance. Why might diversity peak for intermediate disturbance values? (Hint: think of species exhibiting a competition-colonization tradeoff) Can you think of an experiment to test the idea that intermediate levels of disturbance may produce higher levels of biodiversity using these two strains of E. coli? Outline the experimental protocol.