TSP Genotyping
Temperature Switch PCR (TSP)
Purpose
Adapted from Tabone et al. 2009 (2), TSP is a special type of PCR designed for highthroughput screening of single nucleotide polymorphisms (SNPs). It is a useful screen when engineering mutations by two-step allelic exchange, particularly those that do not confer a phenotypic effect, or those for which the phenotypic effect is unknown.
Each reaction contains two primer pairs with fundamentally different melting temperatures (TM): the locus specific (LS_f/LS_r) primers with a relatively high TM, and the nested locus specific (NLS_f/NLS_r) primers with a relatively low TM (Figure 1). In the first part of the PCR the outer LS primers are used to amplify the ~800- 1,000 bp target locus from genomic DNA. Then, a drop in the PCR annealing temperature (hence the name TSP) switches the focus to the inner NLS primers. The NLS_f primer contains the mutant SNP allele at its 3' terminus. This primer only binds to the mutant allele, and thus the 100-200 bp ‘allele specific’ product is only amplified if the mutation is present. If the mutation is absent (i.e. wild type), the NLS_r primer forms a larger, 300-500 bp ‘alternate allele’ product with the LS_f primer. If both mutant and wild type alleles are present (e.g. incomplete purification, mixed population, heterozygotes) then both products should be obtained. It is important to note that regardless of which allele is present, a product should be generated; if no products are obtained, the PCR did not work. This means the chance of missing a rare allele due to PCR failure is minimal.

Figure 1: Outline of primer design for TSP. The LS primers are shown in purple, the LS primers in orange. Figure adapted from (2). (SNP=single nucleotide polymorphism.)
Both NLS primers contain short, non-complementary 5' tails. The purpose of these is to raise the TM of the primers (after several rounds of product amplification at the low annealing temperature). This allows the NLS primers to compete more efficiently with the LS primer pair in the second part of the PCR.
Note: Tabone et al. 2009 (2) developed the method using DNA extracted from the barley plant Hordeum vulgare. GC content differs between (and within) genomes, meaning that some of the lower annealing temperatures may be hard to achieve when designing primers for allelic exchange in other bacteria and/or phage.
Methods
Each method section is supplemented with a tested example: the detection of allele T3380C (T=wild type, C=mutant) in the J gene of bacteriophage λ (NCBI#: NC_001416.1).
Primer Design
The general requirements for TSP primers are as follows (primer TM calculated using IDT oligoanalyzer:
| LS | NLS |
|---|---|
| *Length ~18-28 bp | *Length ~12-18 bp |
| *TM: 60-60°C (aim for 63&Deg;C | *TM core:43-47°C (aim for 45°C) |
| *Product length: 800-1000 bp | *TM with 5' tail: 52-55°C (aim for 53°C) |
| *Product length: 100-200 bp (NLS_f/r); 300-500 bp (NLS_r/LS_f) |
For all primers: use IDT oligoanlayzer (see above) to check for hairpins, self dimers and heterodimers (with the other 3 primers in the mix). Also check final core primers for number of times present in the target genome (once only is desirable).
NLS_f
Begin with the NLS_f primer. This is easiest to place as the 3' end incorporate the SNP. Given the low TM (45°C), this primer will usually be quite short (about 11-16 bp, depending on the GC content of your organism). For efficient amplification, it is desirable for this primer to be ~16 bp, but this is not always achievable due to the low TM; the TM is more important than the length. Once the core region has been determined, add 2-5 non-complementary bp to the 5' end.
Note: the primer can be designed in either direction (3' or 5') from the SNP, as long as the position of the other primers is adjusted accordingly. This allows a little more flexibility when placing this primer (e.g. to avoid mononucleotide runs of >3).
NLS_r
Next, design the NLS_r primer. This should be positioned 100-200 bp downstream of the NLS_f primer, on the opposite DNA strain and running in the opposite direction. It also needs a low TM (45°C), and so again is likely to be short. However, there is more choice for where to position this primer, so avoid repeats and aim for 14-16 bp.
LS_f
The LS_f primer should be positioned ~300-500 bp upstream of the NLS_r primer, on the same strand and in the same direction as the NLS_f primer. Aim for a TM of 63°C.
LS_r
The final primer, LS_r, should be positioned ~800-1,000 bp downstream of the LS_f primer, on the same strand and in the same direction as the NLS_r primer. Aim for a TM of 63°C.
Example: λ J_T3380C
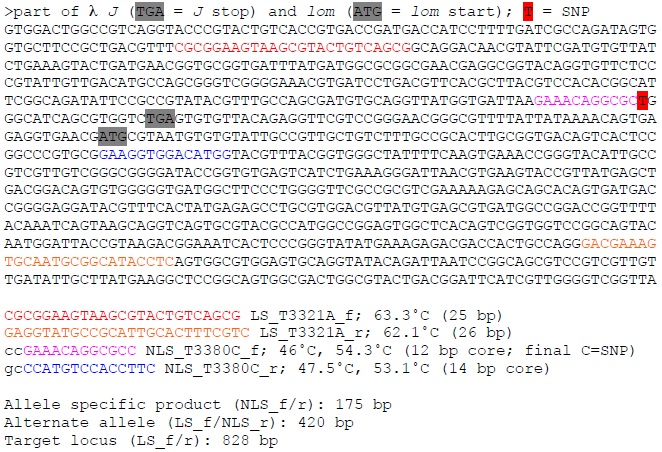
Setting up the PCR
When setting up the PCR, the authors use reaction volumes as low as 5 µl. While this is great when screening large numbers (uses less Taq), it is not practical to pipette such small volumes when screening low numbers. For these situations, a PCR volume of 25 µl has been found to work well. We have tested the protocol with and without combinatorial enhancer solution (CES; (1)), and in many cases CES increases band strength. so we add it routinely (Tabone et al. do not).
Note: NLS primers are present at 5 times the amount of LS primers (final concentrations of 0.5 µM for each NLS primer and 0.1 µM for each LS primer):
| Ingredient | µl | µl |
|---|---|---|
| Water | 3.9 | 9.75 |
| 5xCES | 2.0 | 5.0 |
| 10xPCR buffer | 1.0 | 2.5 |
| dNTPs | 0.2 | 0.5 |
| LS_f (5 pmol/µl; 0.1 µM) | 0.2 | 0.5 |
| LS_r (5 pmol/µl; 0.1 µM) | 0.2 | 0.5 |
| NLS_f (5 pmol/µl; 0.5 µM) | 1.0 | 2.5 |
| NLS_r (5 pmol/µl; 0.5 µM) | 1.0 | 2.5 |
| Taq | 0.05 | 0.25 |
| Genomic DNA (~50 ng/µl), colony re-suspension, culture | 0.25 | 1.0 |
| Total | 10 µl | 25 µl |
Performing the PCR
PCRs should be run on the following programme:
- 95°C for 3 minutes (initial denaturation)
- 95°C for 30 seconds (denaturation)
- 58°C for 30 seconds (LS primer annealing)
- 72°C for 1 minute (extension for LS primers)
- Cycle step 2-4 x 15 (15 cycles to amplify 800-1,000 bp target locus)
- 95°C 10 seconds (denaturation)
- 45°C 30 seconds (NLS core primer annealing)
- Cycle step 6-7 x 5 (5 cycles to amplify the allele specific and/or alternate alleles with the core NLS primers)
- 95°C 10 seconds (denaturation)
- 53°C 30 seconds (NLS + 5' tail annealing)
- 72°C 5 seconds (product extension)
- Cycle step 9-11 x 15 (15 cycles to amplify the allele specific and/or alternate alleles with the NLS primers with 5' tails)
- 4°C 10 minutes (final cooling)
Gel electrophoresis
Once the PCR is complete, run 5-10 µl of each product on a 1.5 % (0.75 g agarose in 50 ml 1 x TBE) gel. Good product separation can be achieved by running at 110 V for 45 minutes. An appropriate DNA ladder for small gene products (e.g. 10 µl of Fermentas exACTGene 100 bp ladder) should be used. Note: sometimes some alternate allele product can amplify with the mutant template (in addition to the allele specific product – i.e. both bands present). However, we have never seen allele specific product with the wild type template. This means there should be no (or at least very few) false positives when searching for a mutant allele.
Example
Here is a gel for which the above instructions were followed for the λ J_T3380C mutation (note: as desired, the 828 bp target locus product cannot be seen due to low numbers of early PCR cycles):

Figure 2: Fermentas exACTGene 100 bp ladder (left) and electrophoresis gel for ! J_T3380C (right). Relevant lanes enclosed by red rectangle, left to right: 175 bp allele specific product (mutant), 420 bp alternate allele product (wild type), both alleles (1:1 mix of templates) & sterile dH2O (-ve control).)
References
- Ralser, M., R. Querfurth, H. J. Warnatz, H. Lehrach, M. L. Yapso, and S. Krobitsch. 2006. An efficient and economic enhancer mix for PCR. Biochem Biophys Res Comm 347:747-751.
- Tabone, T., D. E. Mather, and M. J. Hayden. 2009. Temperature switch PCR (TSP): robust assay design for reliable amplification and genotyping on SNPs. BMC Genomics 10:1-14.
Original protocol by: C. Eshelman Date: 11 Sept 12
Protocol Updated by: ___ Date: ___
Back to RifRamp Protocols